医疗器械出口到马来西亚需要满足一些特定的认证和要求。以下是医疗器械出口马来西亚所需的认证和注意事项:
一、医疗器械立法及主管
马来西亚的医疗器械管理法是《医疗器械管理法(2012)》,该法规的基本框架比较接近美国的医疗器械管理法规。医疗器械的管理部门包括卫生部医疗器械管理局(Medical Device Authority,MDA)和科学技术部原子能许可证局。卫生部医疗器械管理局负责管理除放射性医疗器械和二手医疗器械外的所有医疗器械,而原子能许可证局负责管理放射性医疗器械和二手医疗器械。
二、马来西亚进口医疗器械市场准入流程
(1)马来西亚医疗器械的定义及分类
医疗器械被定义为具备以下用途的任意或联合使用的仪器、器械、器具、机器、植入物、体外试剂或校准器、软件、材料等。它们可以用于诊断、预防、监测、治疗、减轻疾病及损伤,对解剖学或生理过程的研究,支持或维持生命,对器械的消毒,以及通过对取自人体的标本进行体外检查,为医疗或诊断目的提供信息。然而,如果通过药物、免疫等过程发挥这些功能,则不能被定义为医疗器械。根据马来西亚医疗器械管理法规,医疗器械按风险从低到高分为A类、B类、C类和D类四类。其中,A类医疗器械风险最低,B类和C类居中,D类风险最高。
A类产品:低风险,如细胞计数仪。
B类产品:中低风险,如尿液分析仪。
C类产品:中高风险,如心电图仪。
D类产品:高风险,如血液透析机。
(2)体外诊断试剂的分类及注册
体外诊断试剂(in-vitro diagnostic,IVD)是指仅或主要用来为人体提供诊断、监测信息而单独或组合使用的装置,包括试剂、校准器、样品容器等。根据医疗器械管理法,IVD产品根据对个人及公众健康风险影响程度的高低分为A、B、C、D四类。各类别的风险程度和产品举例如下:

对于IVD的注册,需要遵循医疗器械管理法中的专门适用准则,并按照相应的步骤进行注册。

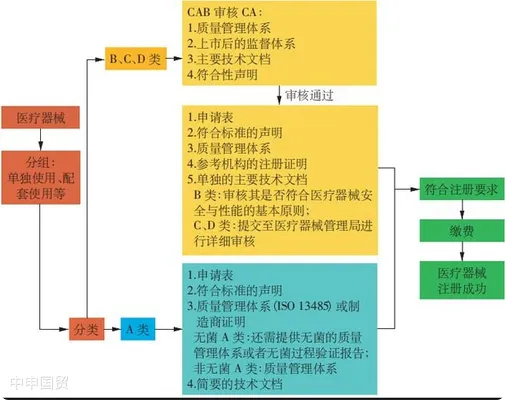
三、马来西亚进口医疗器械注册流程
(1)A类产品
对于A类产品,当地的医疗器械授权代表(Authorized Representative,AR)可以向卫生部医疗器械管理局申请注册,无需MDA授权的合格评定机构(Conformity Assessment Body,CAB)批准。申请人需要提交制造商的ISO 13485证书、测试报告以及标签等文件。
(2)B、C、D类产品
对于B、C、D类产品,当地的医疗器械授权代表需要提交技术报告,并由合格评定机构进行技术文件审查。已经在参考国(如澳大利亚、加拿大、欧盟、日本、美国)获得批准和销售的医疗器械可以通过简化程序进行审核。在审核过程中,需要向合格评定机构提交ISO证书、CE证书等文件。审核通过后,合格评定机构将颁发证书。最终的器械注册文件,包括通用提交档案模板(Common Submission Dossier Template,CSDT)、合格评定机构证书和申请文件,将以电子方式在线提交给卫生部医疗器械管理局进行审查和最终批准。合格评定机构的证书和器械注册证书均应每5年更新一次。
四、注意事项
在医疗器械出口到马来西亚的过程中,需要注意以下事项:
(1)目前马来西亚注册已不再需要自由销售证书(Free Sales Certificate,FSC)。
(2)必须在马来西亚指定的当地代表处提交注册申请。
(3)每个产品只能有一个产品许可证持有人(即持证人)。
(4)产品许可证可以转让给其他持有人。
(5)所有制造商需要获得ISO 13485认证,作为申请注册的必备条件。
总结而言,医疗器械出口到马来西亚需要满足医疗器械管理法规定的注册要求。具体流程包括分类确定、技术文件审查和提交、合格评定机构认证以及最终的在线注册审批。在申请注册之前,应仔细了解相关法规和要求,并确保符合马来西亚的医疗器械出口标准。
最初发布于2023年5月22日 @
标签: 医疗器械进出口




? 2024. All Rights Reserved. 沪ICP备2023007705号-2  沪公网安备31011502009912号
沪公网安备31011502009912号